NUCYNTA ER (tapentadol): THE FIRST AND ONLY OPIOID PROVEN TO TREAT BOTH CHRONIC LOW BACK PAIN (cLBP) AND THE NEUROPATHIC PAIN ASSOCIATED WITH DIABETIC PERIPHERAL NEUROPATHY (DPN)1
IMPROVEMENT IN MEAN cLBP INTENSITY AT WEEK 12 OF MAINTENANCE PERIOD2*
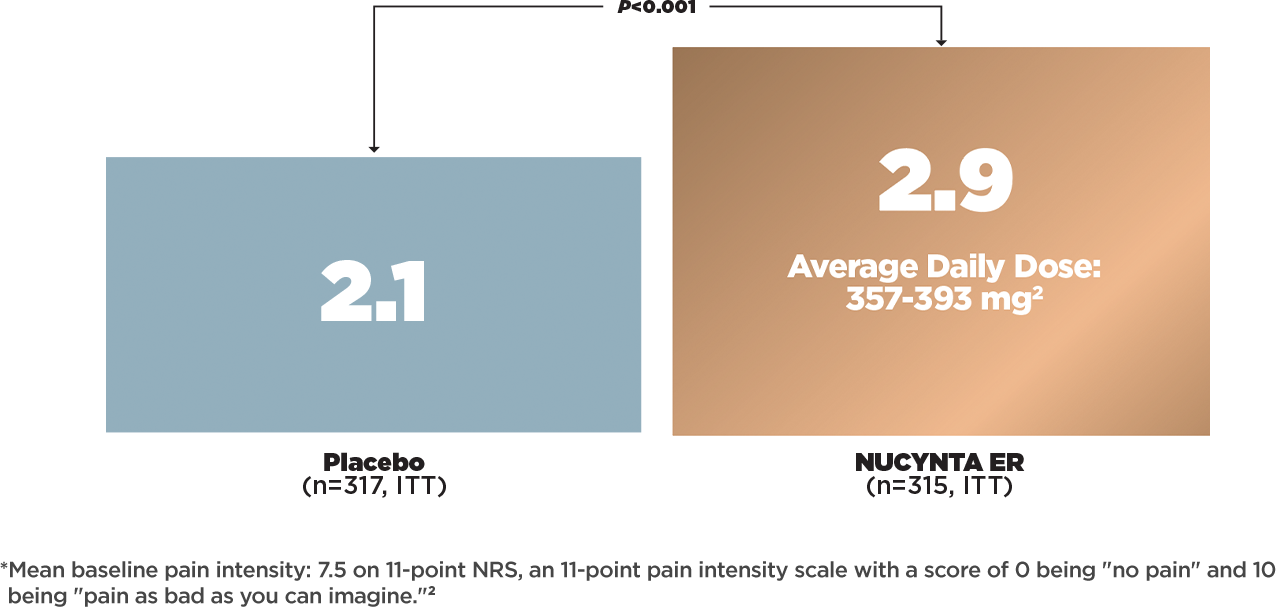
- STUDY DESIGN2
- Prospective, randomized, double‑blind, active‑ and placebo-controlled, multicenter, phase 3 study in patients with moderate to severe chronic low back pain in the US, Canada, and Australia (n=958, ITT)
- Patients were randomized in a 1:1:1 ratio to receive NUCYNTA ER, oxycodone CR, or placebo
- Oxycodone CR (n=326, ITT) with average daily dose of 67‑75 mg was included as active control to confirm assay sensitivity
- PRIMARY EFFICACY ANALYSIS2
- Based on the LOCF imputation method
- Treatment comparisons used the ANCOVA model and were based on LS mean difference from placebo as measured by NRS
- PRIMARY ENDPOINT2
- Change from baseline in mean pain intensity at week 12 of the maintenance period (week 15 of the study), as measured by NRS*
ANCOVA: analysis of covariance; ITT: intent‑to‑treat; LOCF: last observation carried forward; LS: least squares; NRS: numerical rating scale.
Life-Threatening Respiratory Depression:
Serious, life-threatening, or fatal respiratory depression may occur with use of NUCYNTA ER.
NUCYNTA ER: ONE SOURCE OF RELIEF
Watch a clinical presentation on NUCYNTA ER for appropriate patients with chronic low back pain.
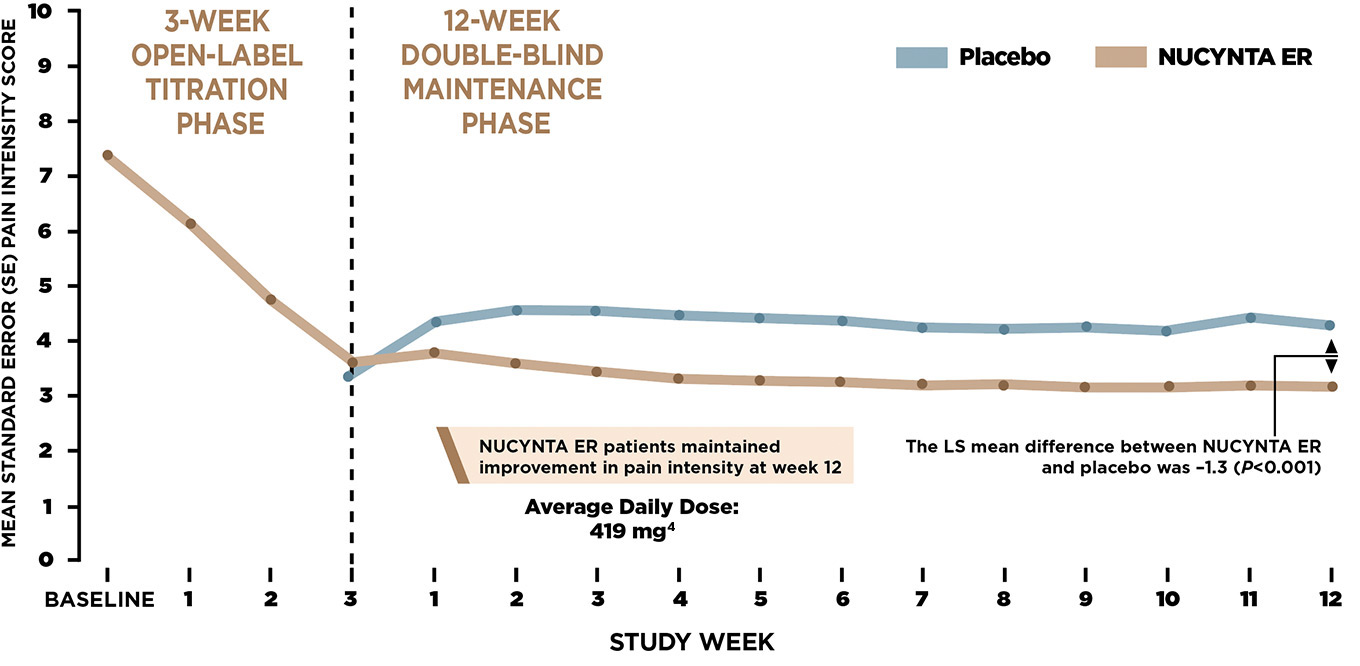
IMPROVEMENT IN MEAN NEUROPATHIC PAIN INTENSITY3
- STUDY DESIGN3
- Double‑blind, parallel‑group, randomized‑withdrawal, placebo‑controlled, multicenter, phase 3 study in patients with painful diabetic peripheral neuropathy
- The trial had two main phases: in the 3‑week open‑label phase, NUCYNTA ER responders (n=389, ITT) received either placebo (n=193, ITT) or NUCYNTA ER (n=196, ITT) 100‑250 mg BID for a 12‑week double‑blind maintenance phase
- PRIMARY EFFICACY ANALYSIS3
- Based on the LOCF imputation method
- Treatment comparisons used the ANCOVA model and were based on LS mean difference from placebo
- PRIMARY ENDPOINT3
- Change from baseline in average pain intensity over the last week (week 12) of the double‑blind maintenance period as measured by the NRS (week 15 of the study; US)
ANCOVA: analysis of covariance; ITT: intent‑to‑treat; LOCF: last observation carried forward; LS: least squares; NRS: numerical rating scale.
Accidental Ingestion:
Accidental ingestion of even one dose of NUCYNTA ER, especially by children, can result in a fatal overdose of tapentadol.
- NUCYNTA ER [package insert]. Stoughton, MA: Collegium Pharmaceutical, Inc.; 2021.
- Buynak R, Shapiro DY, Okamoto A, et al. Efficacy and safety of tapentadol extended release for the management of chronic low back pain: results of a prospective, randomized, double‑blind, placebo‑ and active‑controlled phase Ill study. Expert Opin Pharmacother. 2010;11(11):1787‑1804.
- Schwartz S, Etropolski M, Shapiro DY, et al. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized‑withdrawal, placebo‑controlled trial. Curr Med Res Opin. 2011;27(1):151-162.
- Data on file, Collegium Pharmaceutical, Inc.